| Big Picture | Code | Documentation | Results | Related Tools | Context | User Support |
| Features | Download | Manual | Publications | Pre/Post processing | Authors | Mail list |
| Non-features | SourceForge | Developer guide | Pictures | Pizza.py Toolkit | History | Workshops |
| FAQ | Latest features & bug fixes | Tutorials | Movies | Offsite LAMMPS packages & tools | Funding | User scripts and HowTos |
| Wish list | Unfixed bugs | MD to LAMMPS glossary | Benchmarks | Visualization | Open source | Contribute to LAMMPS |
| . | Pull requests | Commands | Citing LAMMPS | Related modeling codes | . | . |























LAMMPS is a classical molecular dynamics code, and an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator.
LAMMPS has potentials for solid-state materials (metals, semiconductors) and soft matter (biomolecules, polymers) and coarse-grained or mesoscopic systems. It can be used to model atoms or, more generically, as a parallel particle simulator at the atomic, meso, or continuum scale.
LAMMPS runs on single processors or in parallel using message-passing techniques and a spatial-decomposition of the simulation domain. Many of its models have versions that provide accelerated performance on CPUs, GPUs, and Intel Xeon Phis. The code is designed to be easy to modify or extend with new functionality.
LAMMPS is distributed as an open source code under the terms of the GPL. The current version can be downloaded here. Links are also included to older F90/F77 versions. Periodic releases are also available on SourceForge.
LAMMPS is distributed by Sandia National Laboratories, a US Department of Energy laboratory. The main authors of LAMMPS are listed on this page along with contact info and other contributors. Funding for LAMMPS development has come primarily from DOE (OASCR, OBER, ASCI, LDRD, Genomes-to-Life) and is acknowledged here.
The LAMMPS web site is hosted by Sandia, which has this Privacy and Security statement.
 (11/16) Added temper/grem
and fix grem commands to enable tempering
via the generalized replica exchange method (gREM) method.
(10/16) Added a fix
wall/gran/region command which allows
geometric regions to act as boundaries for granular particles.
(9/16) Added options for weighted
load-balancing to the balance and fix
balance commands, which can be useful for better
overall performance of heterogeneous simulation models.
(9/16) Added a fix
cmap command for 5-body CMAP crossterms as defined
by the CHARMM force field between overlapping dihedrals.
(9/16) Added Kokkos support (GPU, Phi)
for long-range electrostatics via the kspace_style
pppm/kk command.
(8/16) Added a fix
controller command to enable guiding of a
simulation to a desired target. If uses a control loop feedback
mechanism known as a proportional-integral-derivative (PID)
controller.
(6/16) Added a Kokkos version of the
ReaxFF potential, i.e. pair_style reax/c/kk, so
it can be run using OpenMP or on GPUs or Intel Phis.
(6/16) Added reactivity extensions to the
USER-DPD package to enable reactive DPD
simulations.
(2/16) Added a dump
custom/vtk command that outputs snapshots in
VTK format readable by the VTK visualization
toolkit or other visualization tools that use it,
such as ParaView.
(2/16) Added a USER-DPD
package for performing DPD simulations at
constant energy/temperature/pressure/enthalpy with an efficient
Shardlow splitting integrator.
(12/15) Significant features added to
LAMMPS in the fourth quarter of 2014 include these new commands:
pair_style mgpt, pair_style
smtbq, pair_style
vashishita, fix
ave/correlate/long, compute
hexorder/atom, compute
orientorder/atom, pair_style
lj/mdf, pair_style lennard/mdf,
pair_style buck/mdf, improper_style
distance, compute chunk/atom bin
sphere/cylinder options, and fix
qeq/fire. See authors here and
details here.
(10/15) Added two new pair styles:
pair_style mgpt for quantum-based model
generalized pseudopotential theory (MGPT) multi-ion potentials, and
pair_style smtbq for second moment tight binding
QEq potentials for ionocovalent bonds in oxides.
(9/15) Significant features added to
LAMMPS in the third quarter of 2014 include a new HTML format for doc
pages with a search option, and these new packages
and commands: USER-DIFFRACTION package,
USER-QTB package, USER-DRUDE
package, USER-SMD
package, COMPRESS
package, USER-TALLY
package, dump h5md,
read_data for multiple data files, pair_style
polymorphic, timer, and a
run_style respa hybrid option. See authors
here and details here.
(8/15) Added a dump
h5md command which can write HDF5 formatted dump
files.
(7/15) Added a USER-SMD
package for performing Smooth Mach
Dynamics, which a SPH-related model applicable to solids.
(7/15) Trying out a new format for the
manual doc pages. Thanks to Richard Berger (JKU)
for scripting restuctured text (rst) and Sphinx tools to do this.
(7/15) Added a USER-DRUDE package which
implements a thermalized Drude dipole model
for polarization effects.
(7/15) Added a USER-QTB package to enable
inclusion of quantum nuclear effects when applicable via 2 commands,
fix qtb and fix qbmsst, as an
extension to classical MD.
(7/15) Added a USER-DIFFRACTION package
to compute virtual X-ray and electron
diffraction patterns.
(7/15) Extended the
read_data command to allow it to be used
repeatedly, e.g. to create a complex system from atoms in multiple
data files.
(6/15) Significant features added to
LAMMPS in the second quarter of 2014 include a new version of the
KOKKOS package and library. See authors
here and details here.
(3/15) Added a PYTHON package with a
python command which embeds the Python interpreter
in LAMMPS and allows Python code you write to be invoked from a LAMMPS
input script, with data passing back and forth between LAMMPS and
Python. Section_python gives an
overview.
(3/15) Significant features added to
LAMMPS in the first quarter of 2014 include these new packages and
commands: PYTHON package with python command to
embed Python in input scripts, CORESHELL
package, pair_style
quip, fix ave/chunk,
compute chunk/atom and other per-chunk
computes, create_bonds, fix
atom/swap, rigid body image
flags, fix gcmc enhancments,
fix ttm/mod, fix tfmc,
pair coul/streitz, fix
rattle, and fix
temp/csld. See authors here
and details here.
(3/15) Added a CORESHELL package with the
capability to run the adiabatic core/shell model for polarization
effects. Section_howto 25 gives an
overview.
(2/15) Added an interface to the
QUIP/libAtoms MD framework framework and its
GAP potentials via a new pair_style quip
command.
(2/15) Added
chunks and per-chunk
computations to the code, as summarized
here.
(1/15) Added a
create_bonds for adding bonds between pairs of
atoms based on a distance criterion.
(1/15) Added a new fix
atom/swap command for performing Monte Carlo
atom swaps (type and charge) either during a dyanmics run or as a
stand-alone MC capability, e.g for surface segregation effects in
alloys.
(11/16) Added temper/grem
and fix grem commands to enable tempering
via the generalized replica exchange method (gREM) method.
(10/16) Added a fix
wall/gran/region command which allows
geometric regions to act as boundaries for granular particles.
(9/16) Added options for weighted
load-balancing to the balance and fix
balance commands, which can be useful for better
overall performance of heterogeneous simulation models.
(9/16) Added a fix
cmap command for 5-body CMAP crossterms as defined
by the CHARMM force field between overlapping dihedrals.
(9/16) Added Kokkos support (GPU, Phi)
for long-range electrostatics via the kspace_style
pppm/kk command.
(8/16) Added a fix
controller command to enable guiding of a
simulation to a desired target. If uses a control loop feedback
mechanism known as a proportional-integral-derivative (PID)
controller.
(6/16) Added a Kokkos version of the
ReaxFF potential, i.e. pair_style reax/c/kk, so
it can be run using OpenMP or on GPUs or Intel Phis.
(6/16) Added reactivity extensions to the
USER-DPD package to enable reactive DPD
simulations.
(2/16) Added a dump
custom/vtk command that outputs snapshots in
VTK format readable by the VTK visualization
toolkit or other visualization tools that use it,
such as ParaView.
(2/16) Added a USER-DPD
package for performing DPD simulations at
constant energy/temperature/pressure/enthalpy with an efficient
Shardlow splitting integrator.
(12/15) Significant features added to
LAMMPS in the fourth quarter of 2014 include these new commands:
pair_style mgpt, pair_style
smtbq, pair_style
vashishita, fix
ave/correlate/long, compute
hexorder/atom, compute
orientorder/atom, pair_style
lj/mdf, pair_style lennard/mdf,
pair_style buck/mdf, improper_style
distance, compute chunk/atom bin
sphere/cylinder options, and fix
qeq/fire. See authors here and
details here.
(10/15) Added two new pair styles:
pair_style mgpt for quantum-based model
generalized pseudopotential theory (MGPT) multi-ion potentials, and
pair_style smtbq for second moment tight binding
QEq potentials for ionocovalent bonds in oxides.
(9/15) Significant features added to
LAMMPS in the third quarter of 2014 include a new HTML format for doc
pages with a search option, and these new packages
and commands: USER-DIFFRACTION package,
USER-QTB package, USER-DRUDE
package, USER-SMD
package, COMPRESS
package, USER-TALLY
package, dump h5md,
read_data for multiple data files, pair_style
polymorphic, timer, and a
run_style respa hybrid option. See authors
here and details here.
(8/15) Added a dump
h5md command which can write HDF5 formatted dump
files.
(7/15) Added a USER-SMD
package for performing Smooth Mach
Dynamics, which a SPH-related model applicable to solids.
(7/15) Trying out a new format for the
manual doc pages. Thanks to Richard Berger (JKU)
for scripting restuctured text (rst) and Sphinx tools to do this.
(7/15) Added a USER-DRUDE package which
implements a thermalized Drude dipole model
for polarization effects.
(7/15) Added a USER-QTB package to enable
inclusion of quantum nuclear effects when applicable via 2 commands,
fix qtb and fix qbmsst, as an
extension to classical MD.
(7/15) Added a USER-DIFFRACTION package
to compute virtual X-ray and electron
diffraction patterns.
(7/15) Extended the
read_data command to allow it to be used
repeatedly, e.g. to create a complex system from atoms in multiple
data files.
(6/15) Significant features added to
LAMMPS in the second quarter of 2014 include a new version of the
KOKKOS package and library. See authors
here and details here.
(3/15) Added a PYTHON package with a
python command which embeds the Python interpreter
in LAMMPS and allows Python code you write to be invoked from a LAMMPS
input script, with data passing back and forth between LAMMPS and
Python. Section_python gives an
overview.
(3/15) Significant features added to
LAMMPS in the first quarter of 2014 include these new packages and
commands: PYTHON package with python command to
embed Python in input scripts, CORESHELL
package, pair_style
quip, fix ave/chunk,
compute chunk/atom and other per-chunk
computes, create_bonds, fix
atom/swap, rigid body image
flags, fix gcmc enhancments,
fix ttm/mod, fix tfmc,
pair coul/streitz, fix
rattle, and fix
temp/csld. See authors here
and details here.
(3/15) Added a CORESHELL package with the
capability to run the adiabatic core/shell model for polarization
effects. Section_howto 25 gives an
overview.
(2/15) Added an interface to the
QUIP/libAtoms MD framework framework and its
GAP potentials via a new pair_style quip
command.
(2/15) Added
chunks and per-chunk
computations to the code, as summarized
here.
(1/15) Added a
create_bonds for adding bonds between pairs of
atoms based on a distance criterion.
(1/15) Added a new fix
atom/swap command for performing Monte Carlo
atom swaps (type and charge) either during a dyanmics run or as a
stand-alone MC capability, e.g for surface segregation effects in
alloys.
This is work by Kirill Lykov (kirill.lykov at usi.ch), Xuejin Li et al at the USI, Switzerland and Brown University, USA to develop new Open Boundary Condition (OBC) methods for particle-based methods suitable to simulate flow of deformable bodies in complex computational domains with several inlets and outlets.
The image (left) and movie (right) show the application of the OBCs to red blood cell flow in a straight pipe, bifurcation, and a part of a capillary network. The program Blender was used for the rendering.
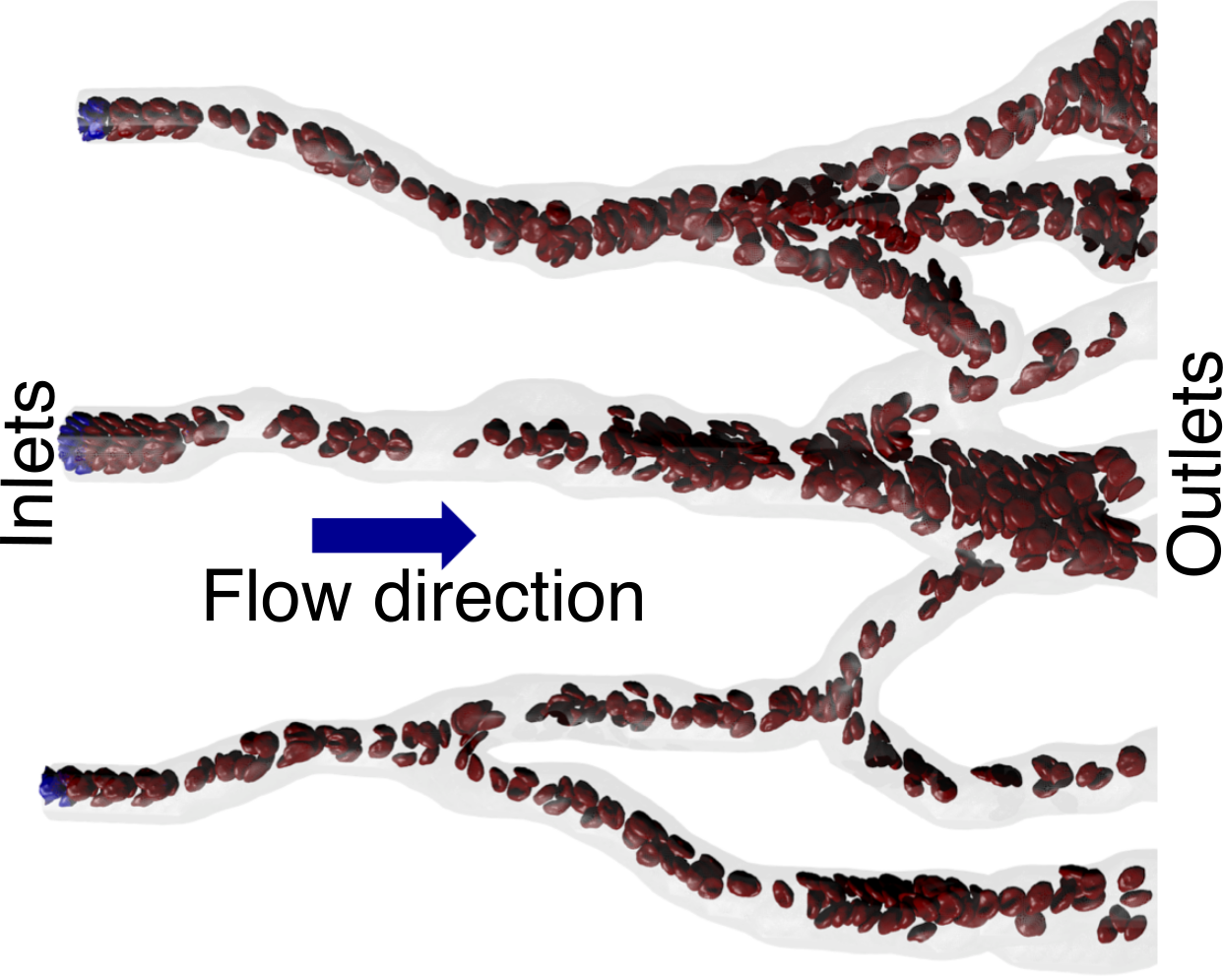

This paper has further details.
Inflow/Outflow Boundary Conditions for Particle-Based Blood Flow Simulations: Application to Arterial Bifurcations and Trees, K. Lykov, X. Li, H. Lei, I. V. Pivkin, G. E. Karniadakis, PLoS Computational Biology 11(8): e1004410 (2015). (doi:10.1371/journal.pcbi.1004410) (abstract)